Two reference insertion variants can be reported at the same position
Taxonomic Profiling sequence list outputs only include reads from the first sequence list input
CLC Network License Manager 5.5.2 temporarily and occasionally fails to serve network licenses
UHGG database for taxonomic profiling contains sequences with long unintentional stretches of N's
PointFinder databases contain incorrect insertions and deletions
Differential Abundance Analysis may use wrong values for samples with multiple metadata associations
Find Best References Using Read Mapping uses only first References and Host references inputs
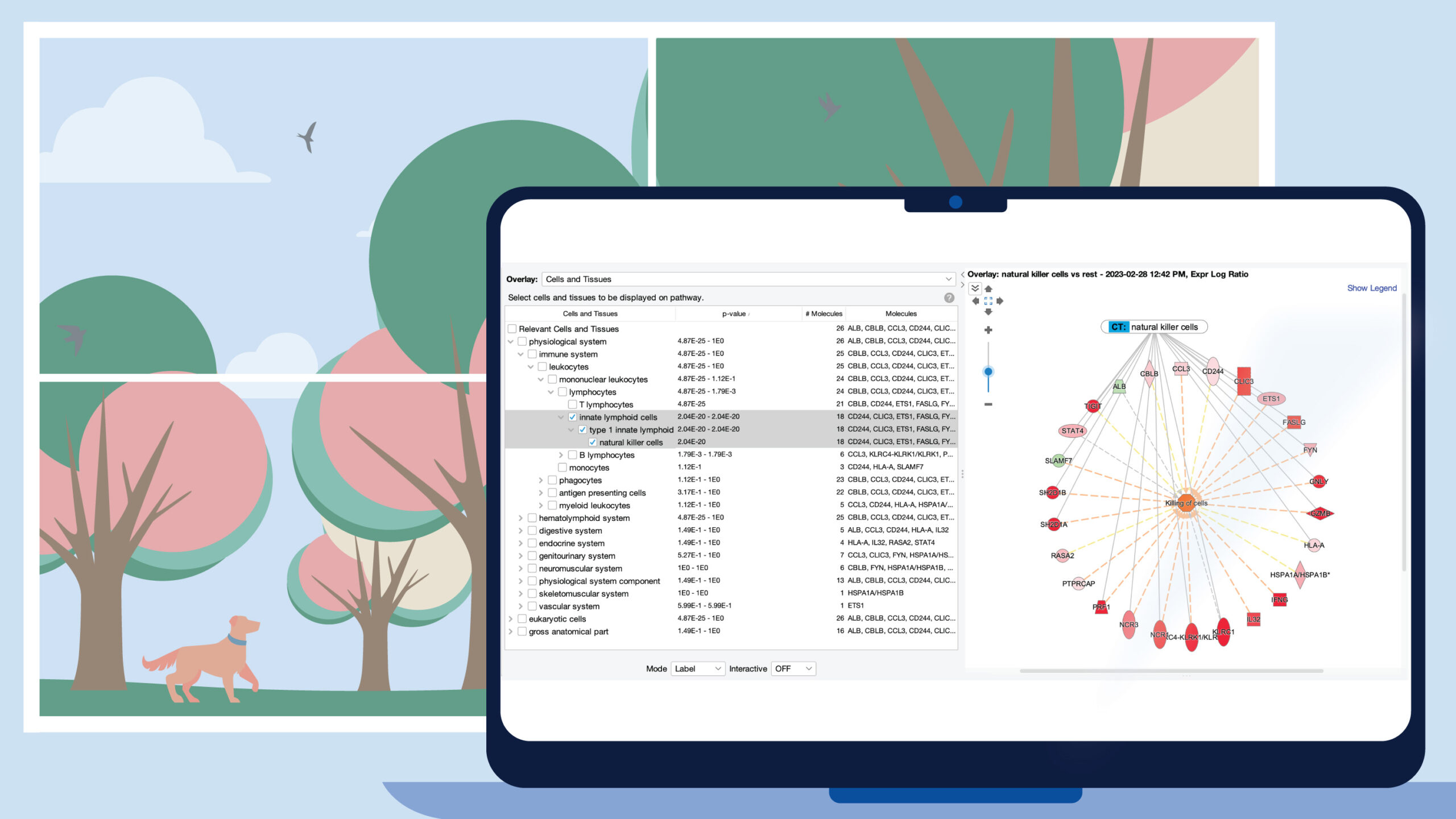
IVA plugin and upload of variants following retirement of Ingenuity Variant Analysis (IVA)
ClinVar from the June 22, 2022 update of CLC reference data is incomplete
Local Realignment can introduce false positive insertions and deletions
QIAGEN RNA-seq Analysis Portal – miRNA expression value downloads may give wrong CPM value
Region and gene-level CNV detection problems when low coverage control targets present
Download Pathogen Database returns too few reference sequences
Transmembrane Helix Prediction always reports no transmembrane regions found
Housekeeping gene normalization is never applied by the tool Differential Expression in Two Groups
Error message appears when Primer info settings are enabled
Error message when working with custom view settings with additional restriction enzymes
Mean reference gene coverage value in QC for RNAScan Panels report is too high
Extract Reads from Selection in abundance tables not working as expected in some circumstances
Taxonomic Profiling accepts multiple host index files but uses only the first one
Taxonomic Profiling reports too few reads in low complexity samples
Import of COSMIC Release v90 is not supported
Restrictions on sequence names when creating BLAST databases
Incorrect annotation and visualization of a small minority of overlapping variants
False positives and misannotation of some fusions in fusion detection workflows
Expression values from UPX 3' Transcriptome Kit data are systematically too high in many cases
Create MLST Scheme tool leads to strains being linked to wrong sequence type in some cases
Incorrect ARO numbers on some ResFinder entries in QMI-AR databases
Bonferroni and FDR multiple testing corrections too strict for differential expression analyses
Local realignment of certain UMI reads may differ between runs
Sporadic corruption of analysis outputs on GPFS file systems
Variants that span a target region are not called
Interquartile range test of the Detect MSI Status tool reports all loci as unstable
Some larger single deletions reported as multiple, individual deletions when affine gap scoring used
RNA-Seq Analysis does not count some reads covering start-end position of a circular chromosome
QIAseq Mitochondria Panel DHS-105Z workflow analyses use incorrect genetic code